Confirming robust genomic associations is only the beginning. These signals serve to plant a flag in a given genomic region, where a haplotype (a linear arrangement of correlated genetic variants) is more often present in disease than in health. However, the physical proximity of the index variant to a protein-coding gene does not imply that this is the gene that, when mutated, gives rise to the phenotype. The variant could be disrupting an enhancer element or another regulatory region for more distant genes (including those that encode microRNAs or long non-coding RNAs, for instance), misleading naïve investigators about the relevant drug target or the gene to disrupt in model organisms. Thus, it is essential that genomic studies be followed by principled searches for the effector transcript that underlies each genetic association.
One potential avenue involves the discovery of coding mutations (either in isolation or in aggregate) that disrupt protein function and phenocopy the original association. For noncoding variants, ancillary information on the pattern of tissue expression of index genes can be found in the GTEx database, which combines expression and human genomic data across many human tissues. This allows one to establish the presence of the transcript of interest in physiologically relevant organs, as well as examine whether noncoding variants associated with the disease phenotype affect message levels (eQTL analysis). Experimental validation that the allelic change leads to the expected perturbation in enhancer or promoter activity is arduous to obtain, but no less crucial in demonstrating causality.
Identifying a likely effector transcript via the above approaches does not by itself establish the direction of effect. At times, variants that change amino acid sequence will have a clear effect on protein function, aligning the direction of the molecular consequence with the disease risk allele. Very often, however, a single amino acid change has no discernible impact, and a search for mutations that alter the protein unambiguously becomes necessary. Through large-scale sequencing approaches, investigators with access to diverse cohorts can identify protein-truncating variants (PTVs) that disrupt protein function (e.g. stop codons, intron-exon splice acceptor sites, frameshifts or read-through mutations), enabling the study of physiological consequences of haploinsufficiency at that site in living humans. If PTVs are statistically more frequent in disease than in health, it can be presumed that their effect on the protein (whether loss of function by deletion of a key activity domain, or gain of function by deletion of an inhibitory domain) is deleterious, and therapies should counteract this effect by either raising the activity or expression of the affected protein (if the PTV induces loss of function) or inhibiting its activity or expression (if the PTV induces gain of function). The reciprocal strategies would be employed if PTVs are found to be protective. Finally, corroborating proof can be obtained by overexpression, silencing or knockout experiments in appropriate cellular or animal model systems, now facilitated by genome editing technologies such as CRISPR/Cas9.
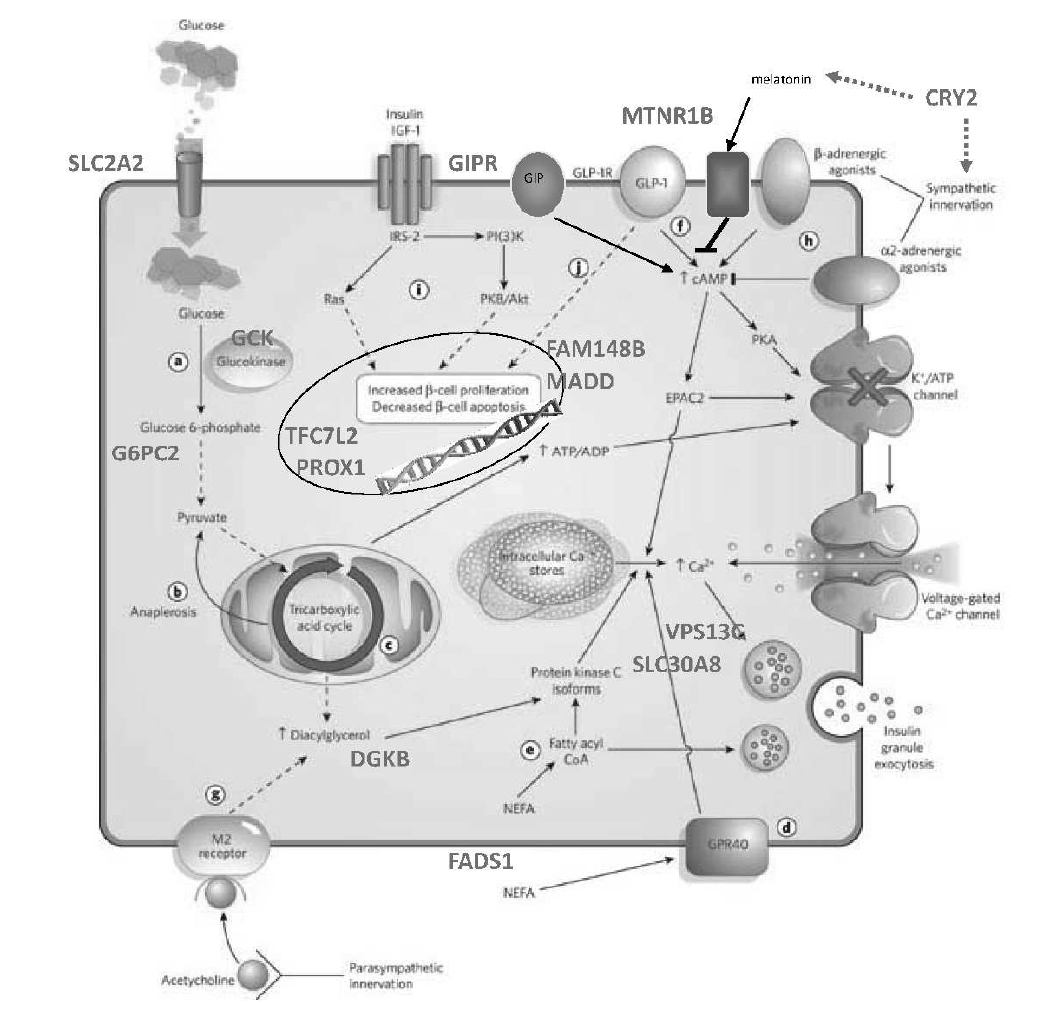
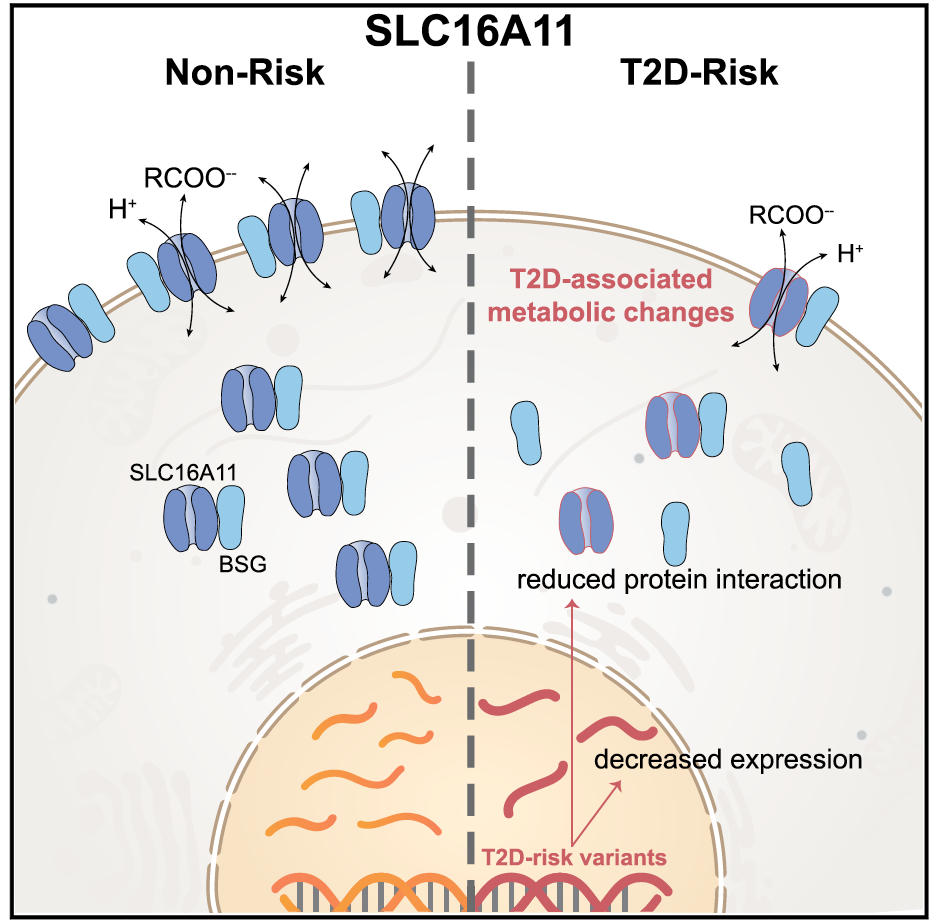
Of the 100+ loci associated with type 2 diabetes or related traits, the Florez lab and collaborators are focusing on the functional characterization of select genes: these include SLC16A11, IGF2, HNF1A, SLC2A2 and ZMPSTE24.
Related Projects
Related Publications